How does changing MCCE’s parameters change accuracy?
MCCE offers a wide range of customizable parameter that influence the accuracy of its pKa predictions. Some major options include varying the internal dielectric constants, numerical Poisson-Boltzmann solvers, Van der Waal functions, conformer sampling level, and explicit crystal water inclusion/removal (implicit/explicit). Each of these options or combinatoric assortments of these options can significantly impact MCCE’s free energy model, and in turn, affects its pKa predictions. In this study, we systematically evaluate how these parameter choices affect MCCE’s calculated pKa values against experimental benchmark values.
Here, we use a set of 36 PDB files sourced from RCSB.org, see BENCHMARK_PROTEINS.tsv. These PDB files were chosen for their varying sizes, as well as the amount of experimental data available for their residue pKa values. Our source for experimental data is the PKAD data set. We have collected a large amount of run data, including pKa data, accuracy, and statistics in this spreadsheet.
Note that the starting and ending residues of protein chains are re-named to NTR and CTR, respectively. At time of writing, MCCE will re-label the first and last residue of a chain, even if the input structure is incomplete.
Accuracy Criteria
Our benchmark dataset consists of 36 PDB files comprising a total of 1,587 residues, of which 425 have experimentally verified pKa values. These will serve as our reference for evaluating the accuracy of MCCE’s pKa predictions. To perform this evaluation, we developed an in-house program that runs parallel MCCE simulations across the 36 structures and compares the predicted pKa values to the experimental values.
We define pKa Accuracy as the percentage of predicted pKa values that fall within ±1 pH unit of the corresponding experimental values. We report the root-mean-square deviation (RMSD), noting that it is sensitive to outliers; where appropriate, we assess how removing outliers affects this metric. The higher the pKa Accuracy is, and the lower the RMSD is, the better the run.
The data presented below were generated using the NGPB Poisson–Boltzmann solver with an internal dielectric constant of 8 and our modified, unscaled van der Waals function. Under these conditions, we observed a pKa Accuracy of 82.35% and an RMSD of 0.88.
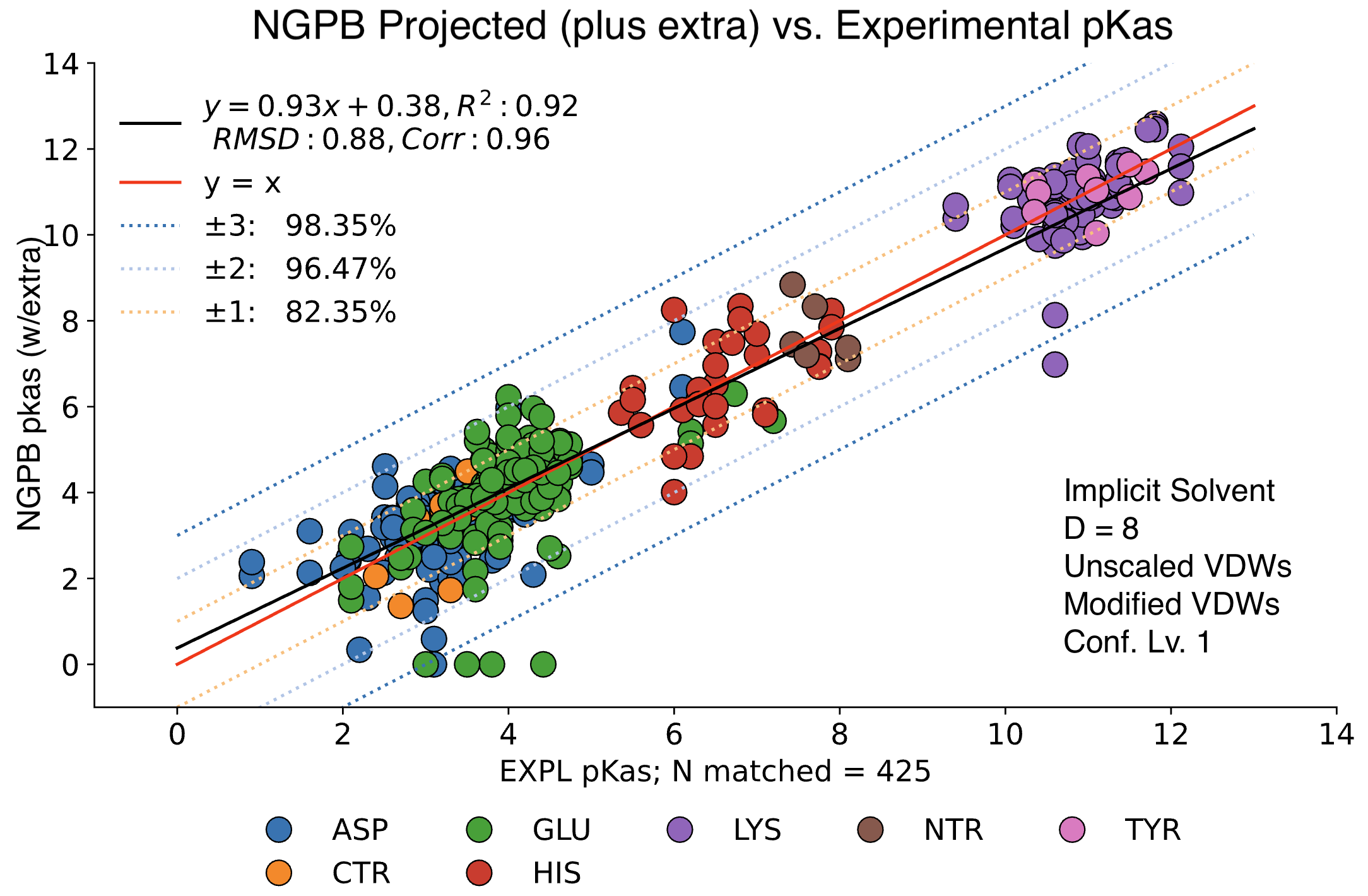
In this graph the experimental pKa values are on the X-axis and the MCCE-predicted pKa values on the Y-axis. This particular run incorporates extra values, which are residue-type-specific adjustments applied after the initial pKa calculations. These adjustments shift all predicted pKas of a given residue type uniformly up or down to reduce systematic bias. However, it’s important to note that while extra values can correct for bias, they do not address the variance within a residue group relative to experimental values.
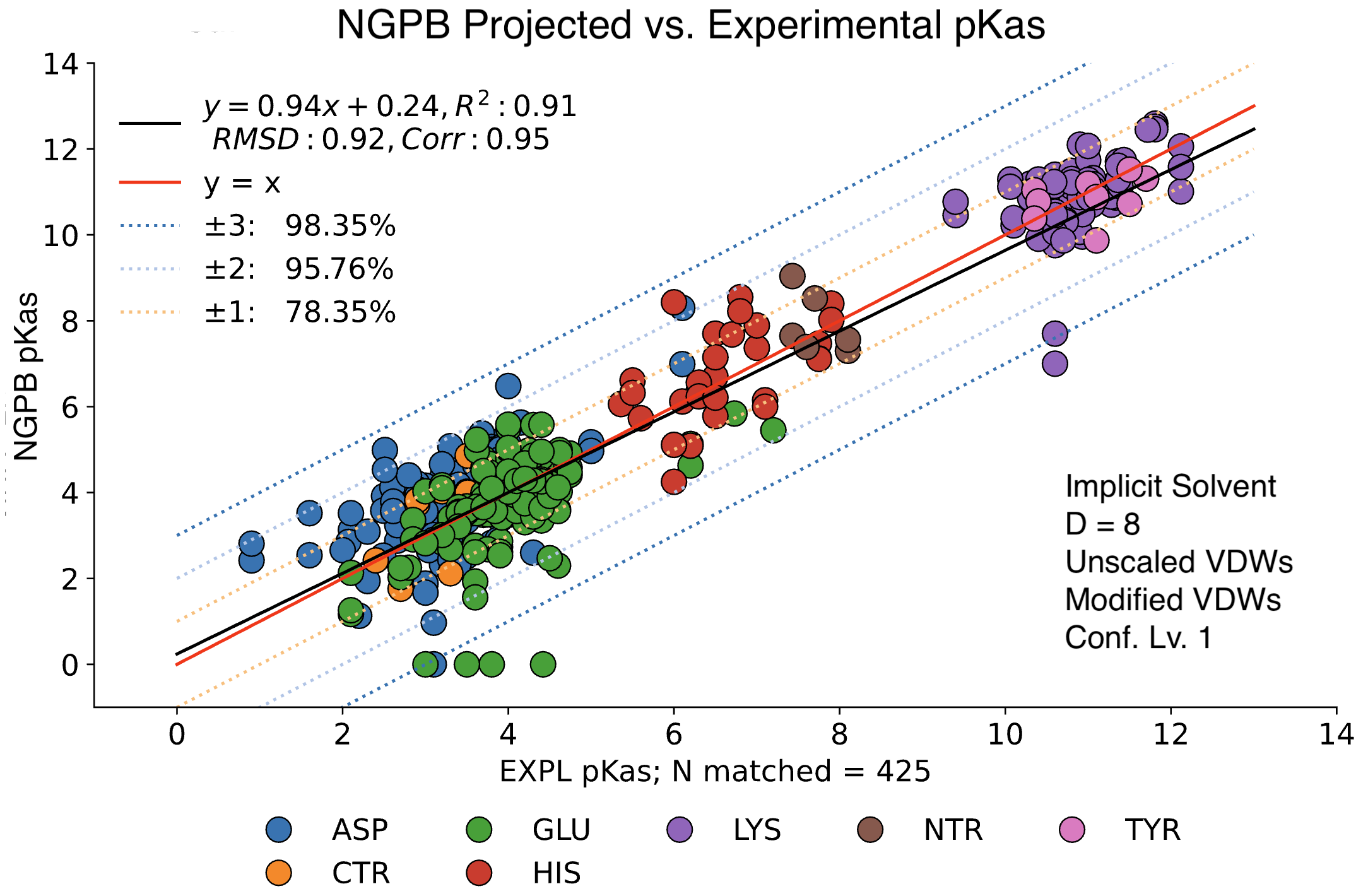
For comparison, the following graph shows the same dataset without applying extra values. Here, we observe the pKa accuracy was reduced by 4% to 78.35%, and RMSD was inflated to 0.92.
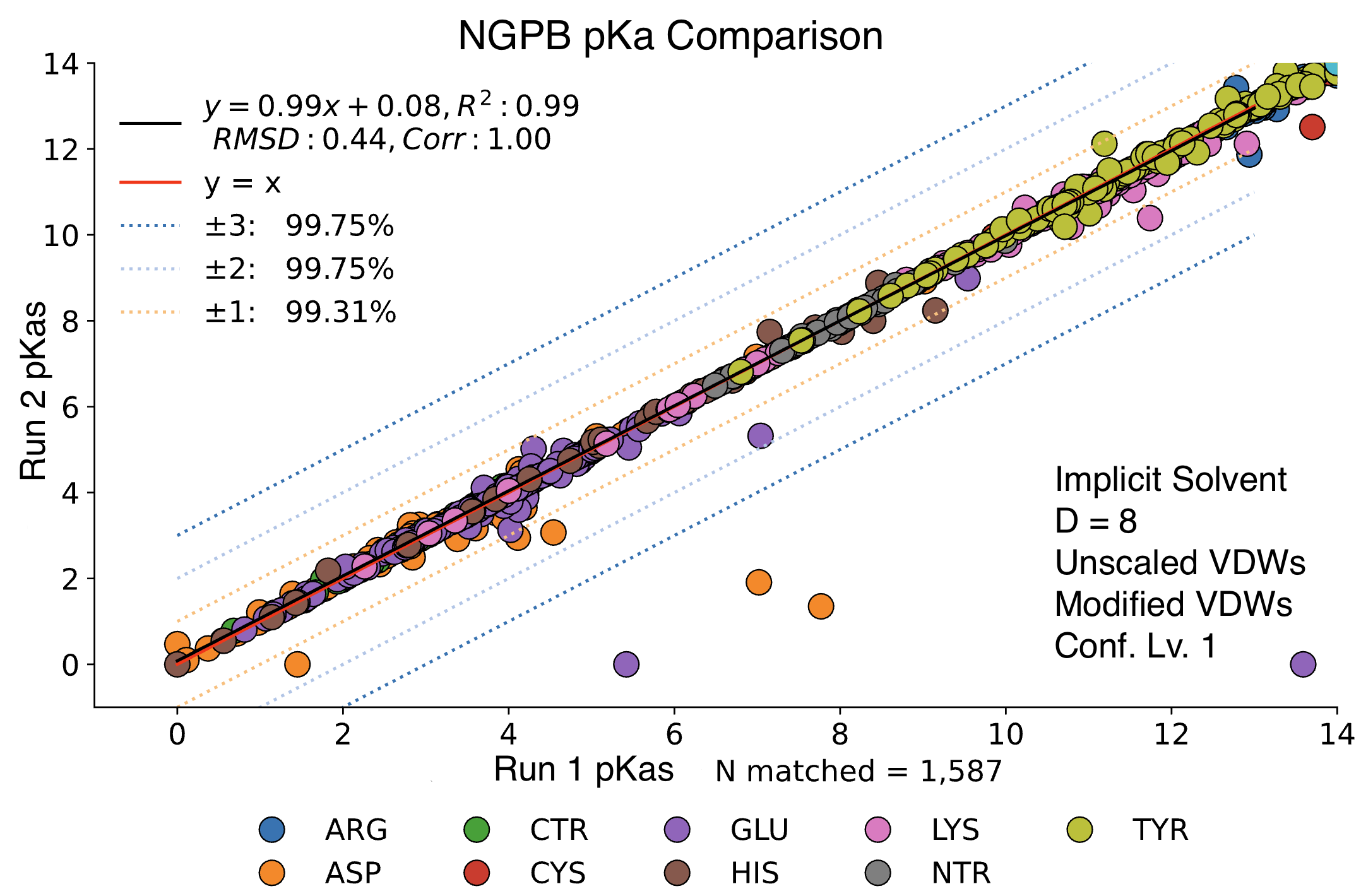
Stochastic Variation Between Runs
Steps 2 and 4 of MCCE incorporate randomness. For step 2, MCCE keeps rotamers that pass energy clash and steric filters, using a randomized exploration of conformational space. Due to the energy minimization, it’s typical that very similar conformers are kept between two different runs. Step 4 features Monte Carlo calculations where millions of random conformers are proposed, and moves are accepted or rejected using the Metropolis criterion. Millions of microstates are accepted, so it’s expected to see minimal random variation between runs. Below we see a direct comparison of two NGPB runs with identical settings. The vast majority of pKas are practically identical, though a few notable exceptions have failed to converge. While random chance does play a role in the final pKa calculation, most of the differences we will observe in future comparisons can be explained by differences in parameter choice.
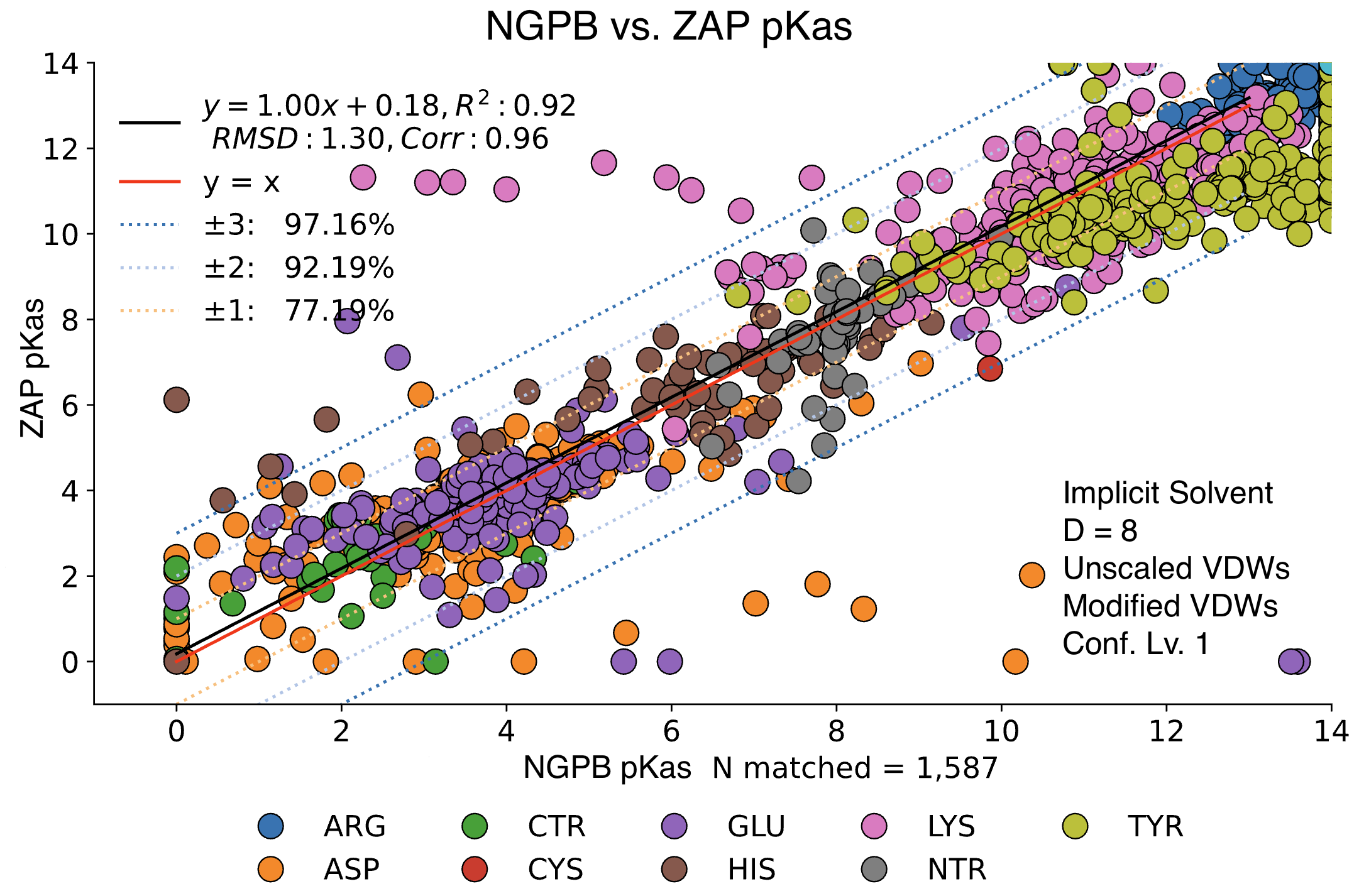
Poisson-Boltzman Solvers
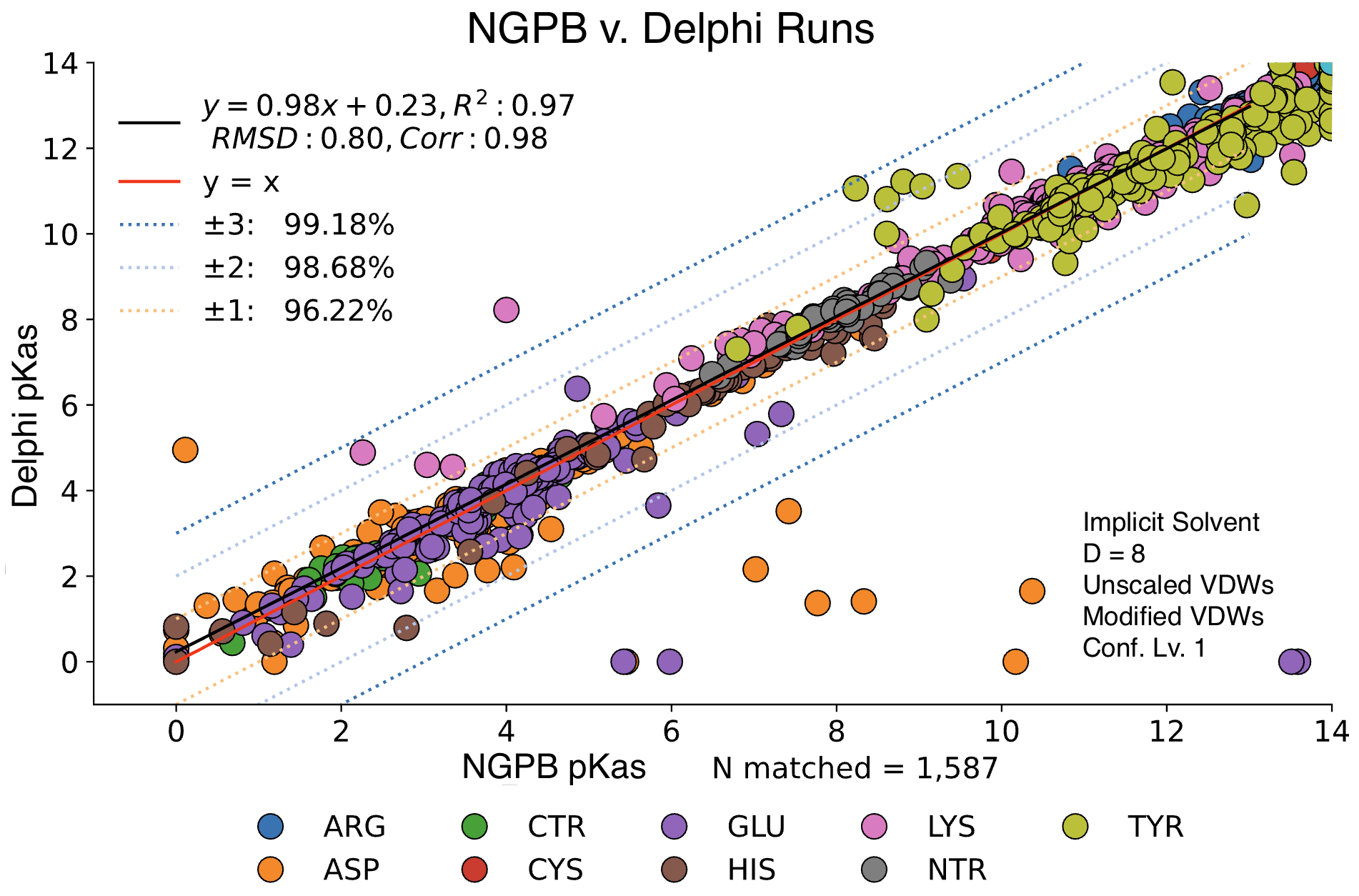
MCCE4 is currently capable of switching between three different Poisson-Boltzmann solvers: ZAP (OpenEye), Delphi, and Next-Generation Poisson Boltzmann (NGPB created by partners in the CONCEPT Lab @ Istituto Italiano di Tecnologia (ITT) in conjunction with Gunner Lab).
Here, we examine the pKa accuracy of our three solvers on our testing set. These are implicit solvent runs, using ε=8, and modified unscaled VDW equations (see below). We have removed all “extra” terms for these graphs, so we are only seeing the solvers and MCCE’s calculations in the final output. This highlights the consistency of MCCE between the different solvers. ZAP is typically the fastest solver, however as it requires a special license from OpenEye, we anticipate NGPB will be support most users’ pKa calculation needs.
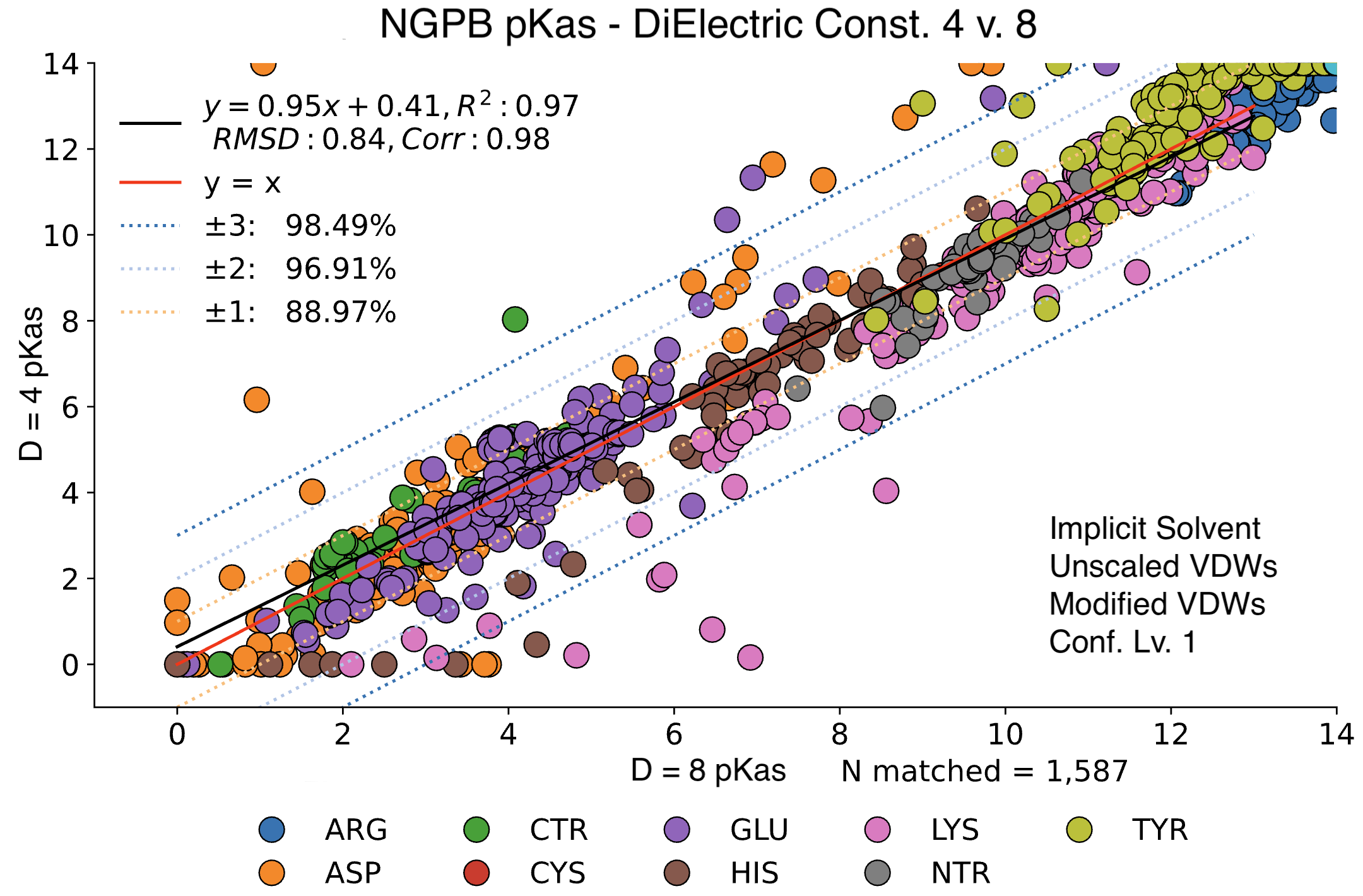
Impact of Dielectric Constant ε
MCCE output shows significant variance depending on the inner dielectric constant (ε).
Below, we compare the optimized results from two dry NGPB runs: the left with ε=4 , and the right ε=8. Both runs use unscaled modified Van der Waals equations. The different residue groups are distributed similarly, but the ε=4 runs experience a greater variance, especially for the 425 residues with experimental data.
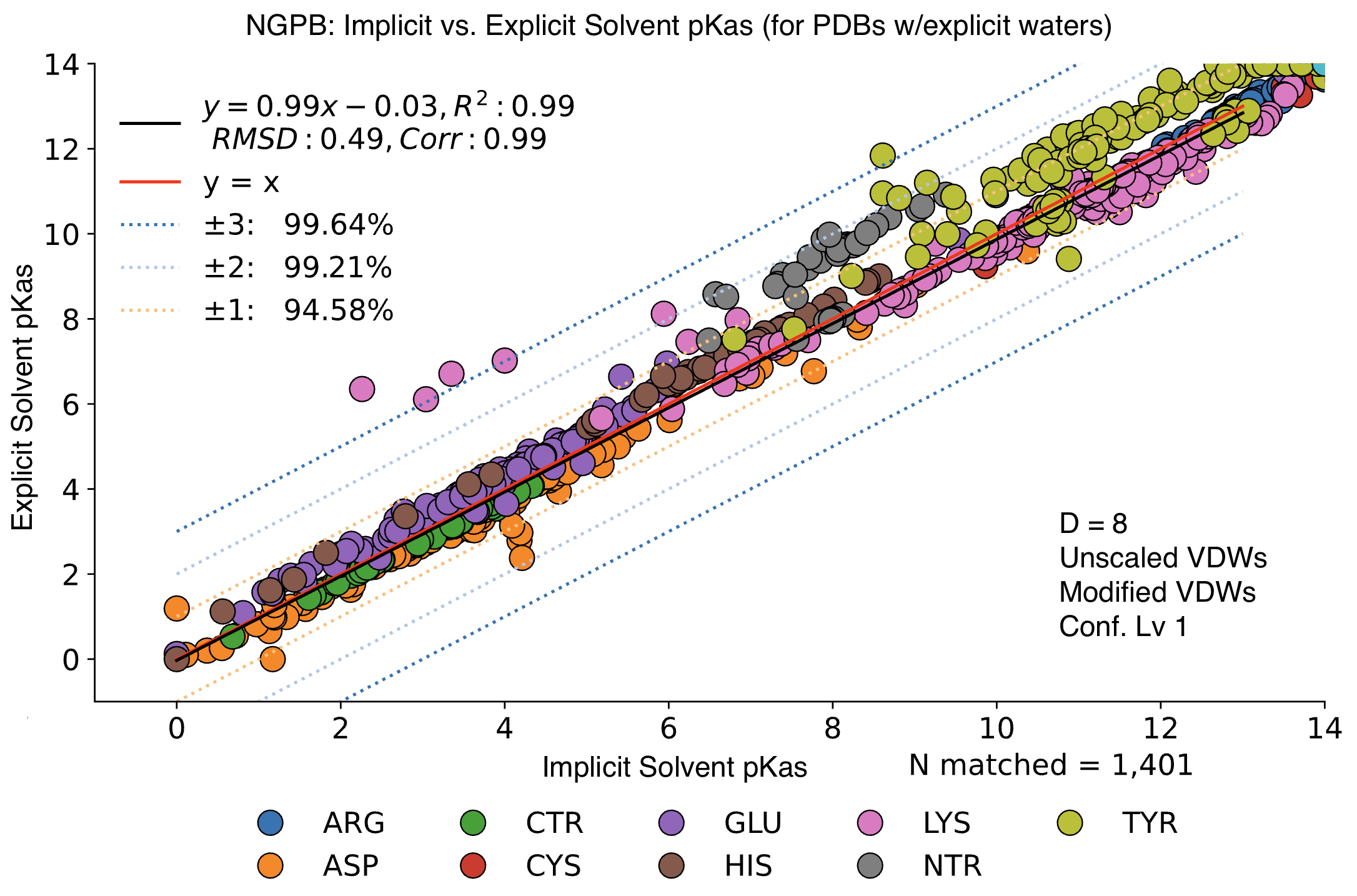
Explicit vs. Implicit Solvent Runs
We observed little change in pKa accuracy when waters are removed from the input PDB. If no explicit waters are present in a PDB file, MCCE will assume an implicit solvent and run accordingly. Implicit runs enjoy slightly better accuracy vs. explicit runs.
In this graph, we see minimal changes in pKa calculation between wet and dry runs of NGPB for ε=8. Note that this is simply a strict comparison of calculations for across all residues between a wet and dry run, not just the experimental data. Additionally, while other comparisons deal with our total benchmark set, the following graph includes only PDB files containing explicit waters. If –dry is selected as an option, these explicit waters will be deleted, and an implicit solvent used. Between the 27 PDB files referenced, there were 3787 waters.
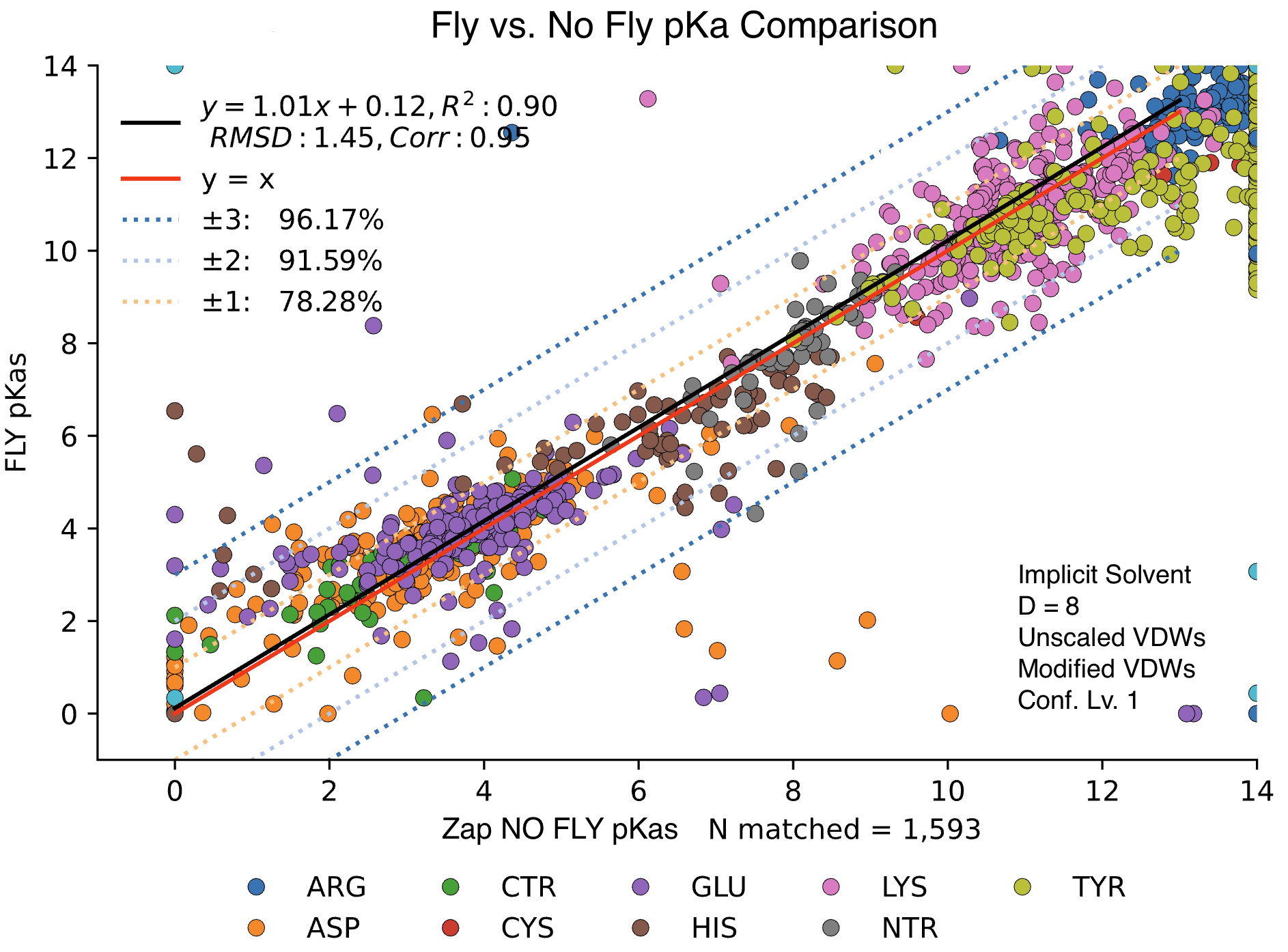
Impact of On-The-Fly Computations
Another possible option, for step 3 of MCCE, is “–fly”. Typically, desolvation energy is computed by performing a single Poisson-Boltzmann Equation run in the mixed dielectric environment and subtracting a precomputed reaction field energy value. However, this approach becomes inaccurate when the side chain conformation changes and partial charges- especially on surface Hydrogen atoms- shift, since the precomputed reference energy (stored in the ftpl file) is fixed, and does not account for such conformational variation.
The “–fly” flag improves this by computing the reference reaction field energy dynamically, or “on-the-fly”. For each conformer, both the reference and in-protein reaction field energies are calculated freshly. The desolvation energy is then obtained as the difference between these two values, making the result less sensitive to changes in conformer orientation.
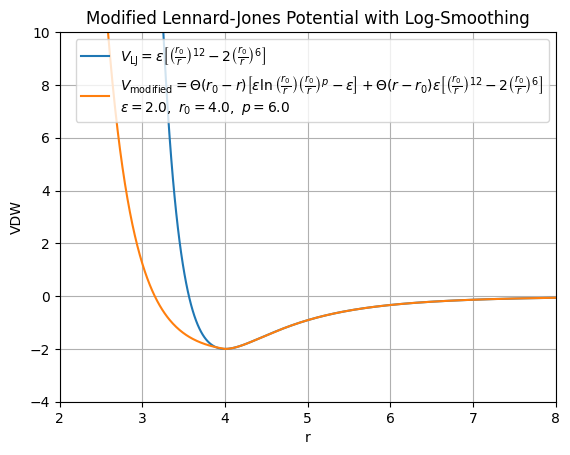
Modified Van-der-Waals vs. Traditional Van-del-Waals
The traditional van der Waals (Lennard-Jones) potential for modeling interactions between atoms is given by (UNDER CONSTRUCTION…):



Conclusions
In conclusion, we demonstrate that for the used Benchmark PDB files, MCCE represents a robust way to estimate pKa values, showcasing consistency between solvers and other settings and conditions. ZAP remains the fastest option, but for those without a license, NGPB provides good accuracy. For most pKa findings needs, we recommend runs that use options dry, ε = 8, conformer sampling Lvl. 1, and NGPB. If this is not sufficient, try conformer sampling Lvl. 2. Using the –fly tag on step 3, for dynamic reference reaction field energy computation, can also reduce variance in the final calculation.
Special thanks to the National Science Foundation, the Department of Energy, and the City College of New York for making this research possible.